Projects
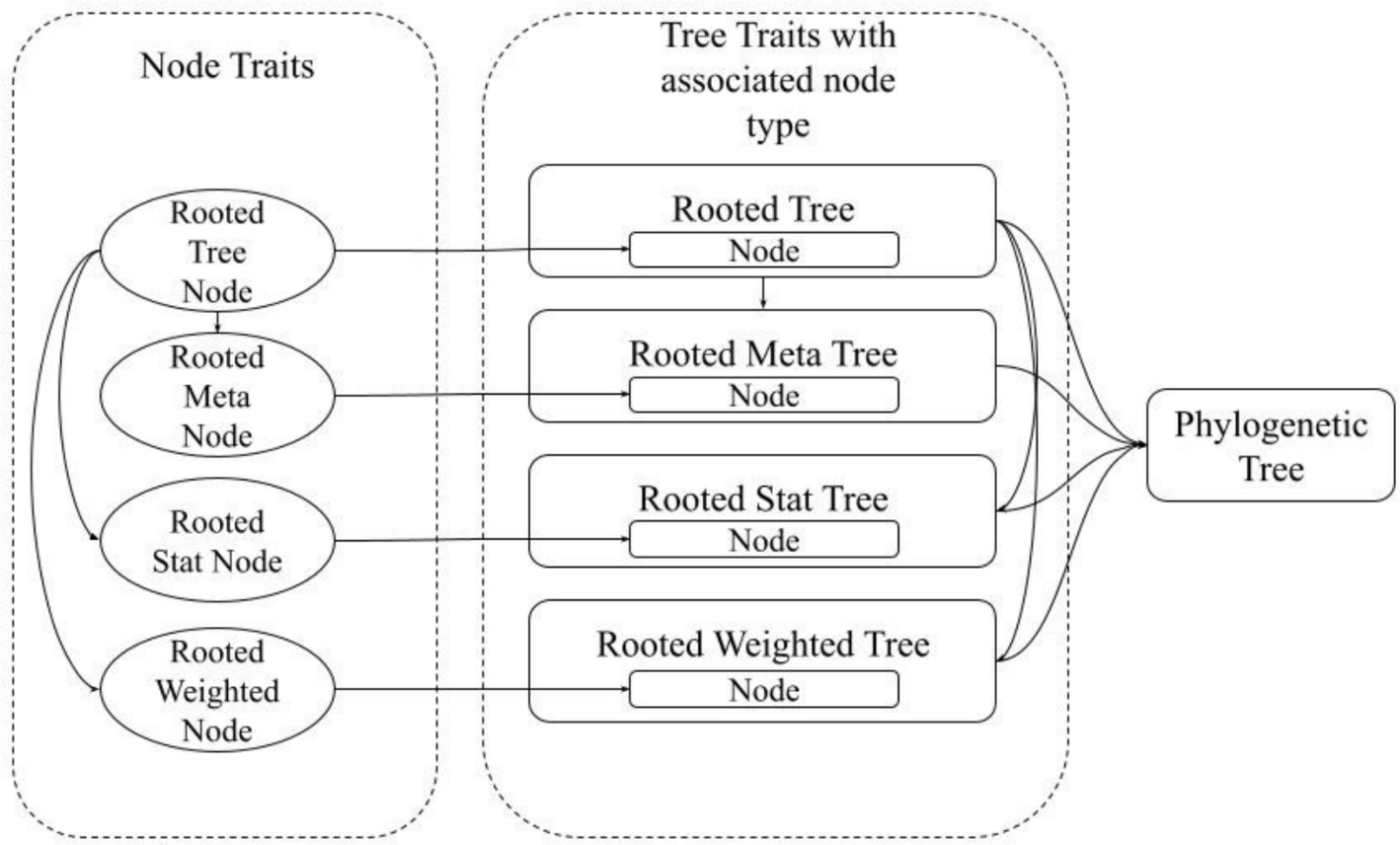
Phylo-rs
Phylo is a fast, extensible, general-purpose, and WebAssembly-capable library for phylogenetic analysis and inference written in Rust. Phylo-rs leverages a combination of memory safety, speed, and native WebAssembly support offered by Rust to provide a robust set of memory-efficient data structures with basic algorithms ranging from tree manipulation with SPR to computing tree statistics such as phylogenetic diversity.
ml-seqclass
Ml-seqclass is a tool that aligns reads to references by first building a suffix array, then aligning the reads to the references by finding seed matches and extending them. After finding all alignments, an EM algorithm is employed to identify the most-likely true source for each read, and to estimate population proportions of the detected references.
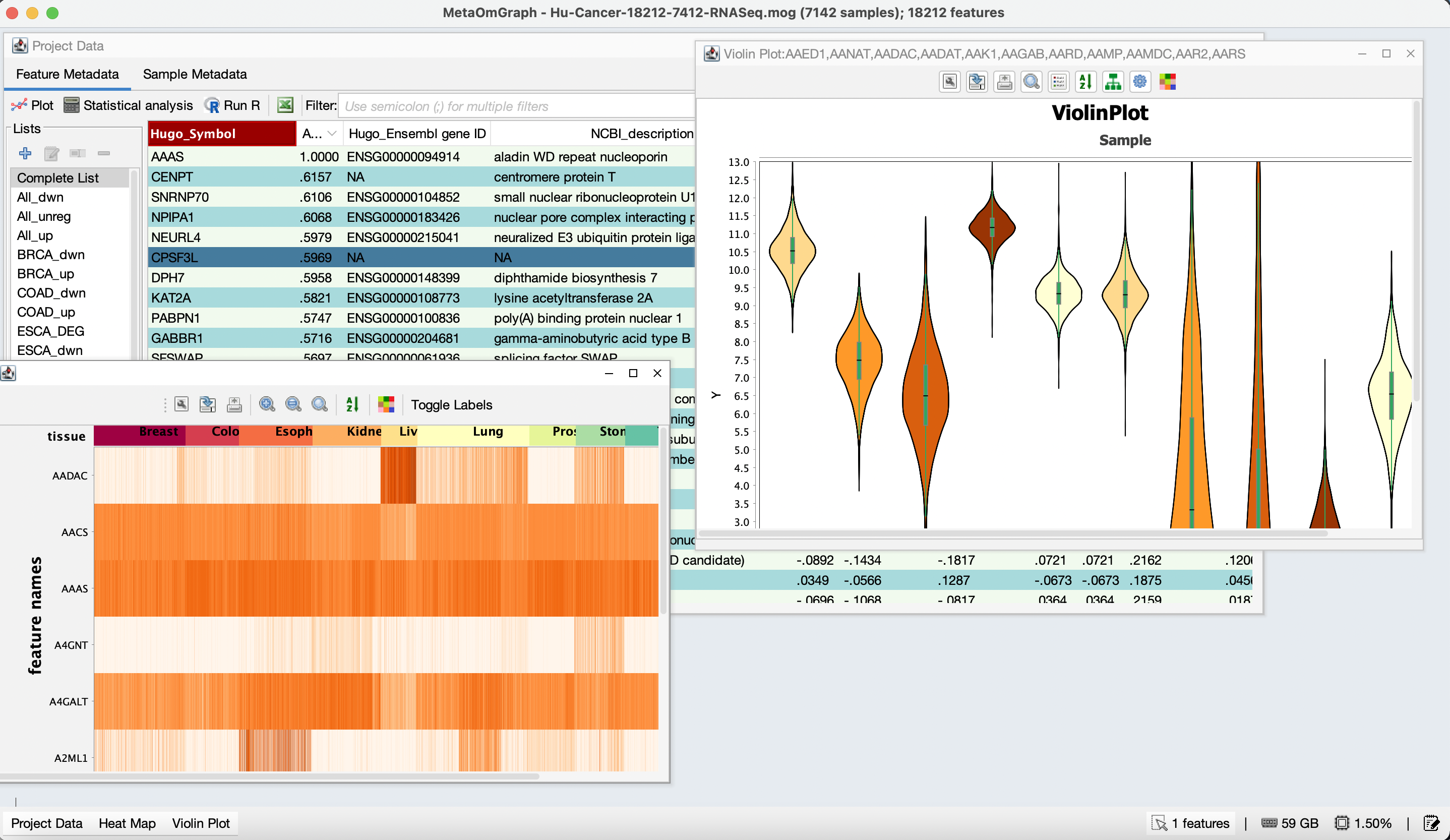
MetaOmGraph
MetaOmGraph (MOG) is a Java software to interactively explore and visualize large datasets. MOG is user-centered software, designed for all types of data, but specialized for expression data. It combines the ability to analyze very large data sets in real time with metadata analysis, statistical analysis, list-making, and graphing capabilities.
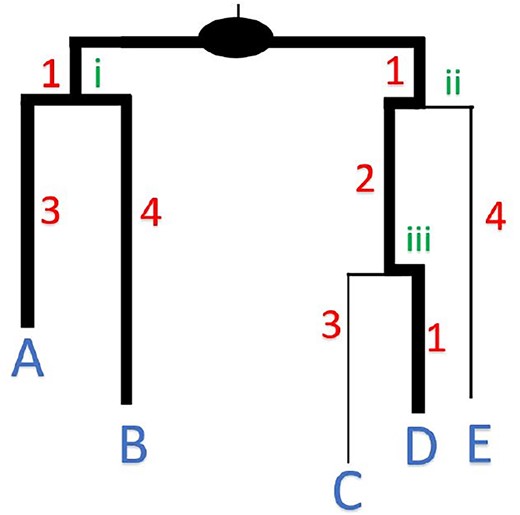
PD Stats
PD Stats is a project designed with algorithms to compute Phylogenetic Diversity (PD) Statistics - Minimum, Maximum, Average, Variance on a phylogenetic tree.

iGTP
iGTP is a software package for phylogenetic analyses using gene tree parsimony (GTP).
DupTree
DupTree is a program for phylogenetic analyses using gene tree parsimony. That is, given a collection of binary gene trees, DupTree searches for a species supertree that implies the fewest number of gene duplication events.
RF-Supertrees
This program takes as input a collection of rooted trees and heuristically searches for a binary supertree that minimizes the total (rooted) Robinson-Foulds distance between the supertree and the input trees.
biclique
This is an implementation of maximal biclique enumeration algorithm from Alexe et al. (http://citeseer.nj.nec.com/alexe02consensus.html)
News